|
EXAMINING THE FUNCTION OF
PROTEINS AND PROTEIN NETWORKS WITH THE YEAST TWO-HYBRID
SYSTEM
Russ Finley, April 1997
I. SUMMARY
The yeast two-hybrid system provides a relatively straight forward
approach to understanding protein function. Section
II outlines the basic components of the
interaction trap, a yeast two-hybrid system developed in the Brent
lab (Gyuris et al., 1993). More detailed background information can
be obtained in a number of recent reviews (Ausubel et al., 1987-1996;
Finley and Brent, 1995; Mendelsohn and Brent, 1994). Section
III contains an interactor hunt protocol,
which is a condensed and updated version of the original
protocol we first posted on the Internet in 1992 and subsequently
updated (Finley and Brent, 1995; Finley et al., 1997). The version
presented here is the one we currently use in our lab and represents
our attempts to streamline and scale up the these techniques to
facilitate characterization of large networks of interacting
proteins. It is also useful for individual hunts. Section
IV discusses alternative approaches
specifically designed to look at large protein networks; the ultimate
goal of developing these and related approaches is to eventually map
all of the interactions encoded by a genome. Section
V discusses briefly two-hybrid approaches
to understanding the functions of individual protein
interactions.
II.
INTRODUCTION
Several different two-hybrid systems have been developed to study
protein function. The garden-variety application is to learn about
the function of a given protein by isolating proteins that interact
with it, usually by screening a cDNA library. To conduct such an
interactor hunt, a protein is expressed in yeast as a fusion to the
DNA-binding domain of a transcription factor lacking a transcription
activation domain. The DNA-binding fusion protein is generally called
the bait . The yeast strain also contains one or more
reporter genes with binding sites for the DNA-binding domain. To
identify proteins that interact with the bait, a plasmid library that
expresses cDNA-encoded proteins fused to a transcription activation
domain is introduced into the strain. Interaction of a cDNA-encoded
protein with the bait results in activation of the reporter genes,
allowing cells containing the interactors to be identified.
The two-hybrid system developed in the Brent lab (the interaction
trap) uses the E.coli protein LexA as the DNA-binding domain
and a protein encoded by random E. coli sequences, the B42
"acid blob", as the transcription activation domain. Both proteins
are expressed from multicopy (2µ) plasmids; the LexA fusion, or
bait, is expressed from a plasmid containing the HIS3 marker,
and the activation domain fused protein, sometimes called the
prey, is expressed from a plasmid containing the
TRP1 marker. In the most commonly used bait plasmid, pEG202,
the bait is expressed from the constitutive yeast ADH1
promoter. Related bait plasmids are available that express the bait
fused to a nuclear localization signal. The most commonly used prey
plasmid, pJG4-5, expresses proteins fused to the B42 activation
domain, the SV40 nuclear localization signal, and an epitope tag
derived from hemagglutinin, all driven by the yeast GAL1
promoter which is active only in yeast grown on galactose. Use of the
GAL1 promoter to express the prey allows toxic proteins to be
expressed transiently and helps eliminate many false positives in
interactor hunts. The interaction trap uses two reporter genes that
carry upstream LexA binding sites or operators: LEU2 and
lacZ. The LEU2 reporters are integrated into the yeast
genome; the lacZ reporters typically reside on 2µ
plasmids bearing the URA3 marker, though integrated versions
are also available. Several versions of the LEU2 and
lacZ reporters exists that have a range of sensitivities based
on the number of upstream LexA operators. In general the LEU2
reporters are more sensitive to a given interacting pair of proteins
than the lacZ reporters (Estojak et al., 1995); however,
highly sensitive lacZ reporters have been used that contain
several LexA operators and transcription terminator sequences
downstream of the lacZ gene (S. Hanes, personal
communication).
More details about the different strains and plasmids available
for the interaction trap can be found elsewhere (Ausubel et al.,
1987-1996; Brent et al., 1997; Estojak et al., 1995; Finley and
Brent, 1994; Finley and Brent, 1995; Finley et al., 1997; Gyuris et
al., 1993)
III.
INTERACTOR HUNT PROTOCOL
Below I refer to typical strains and reporters needed for an
interactor hunt. These include the sensitive LEU2 reporter strain
EGY48, the sensitive lacZ reporter plasmid pSH18-34, a plasmid to
express LexA fusions such as pEG202, and the library plasmid pJG4-5.
The following is a condensed version of a previously published
protocol (Finley and Brent, 1995). It is intended to clarify and
expand on some important points in the original protocol. More
details can be found at the web sites (Brent et al., 1997; Finley et
al., 1997)
A. Testing baits Part 1: Does the bait activate
transcription?
Before performing an interactor hunt it is very important to know
the level of background activation by the bait protein itself. Almost
every LexA fusion will activate the LEU2 reporter in EGY48 to some
extent by itself. The amount of activation by a bait determines how,
and whether, an interactor hunt is done. The most useful way to
measure the level of activation is to determine the fraction of
living cells that are able to grow in the absence of leucine (on leu-
plates). Although it is not immediately obvious why a more strongly
activating bait allows a larger fraction of EGY48 cells to grow in
the absence of leucine, determination of this fraction is essential
to performing an interactor hunt. The fraction can be represented as
the number of colonies that grow on a leu- plate (Leu+ colonies) per
living yeast cell plated. The number of living cells, or colony
forming units (CFU), in an aliquot of cells is determined by plating
dilutions on plates that contain leucine. Thus, the frequency of Leu+
colonies (or Leu+/CFU) is a ratio of the number of colonies that form
on leu- plates over the number that form on plates that contain
leucine. The test is done with the selection strain (the
strain that already contains the lacZ reporter and bait plasmids)
which is transformed with the empty library plasmid, pJG4-5; this
closely mimics the conditions under which the selection for
interactors will ultimately be performed. For a bait that is
virtually unable to activate the LEU2 gene by itself, the frequency
of Leu+ colonies in the test will be less than
10-6 (i.e., less than 1 Leu+ colony
will form when 106 CFU are plated
on the leu- plates). Baits that activate a moderate level of
transcription will result in Leu+ colonies at frequencies from
10-4 to
10-5.
It is important to plate at least
106 CFU onto the leu- plates when
testing a bait for activation of LEU2. To screen a typical library of
106 individual cDNAs, it will be
necessary to plate over 106 CFU of
the selection strain transformed with the library onto the leu-
plates to select for interactors. If the background activation by a
bait were tested by plating only
103 or
104 CFU onto leu- plates, and only
one or a few Leu+ colonies form, it would be tempting to conclude
that the bait activates LEU2 at a sufficiently low level to be used
for an interactor hunt. However, if one were to then attempt to
thoroughly screen a library of 3 x
106 individual cDNAs by plating
over 3 x 106 CFU onto the leu-
selection plates, at least 3000 colonies would form; these would all
be expected to be false positive (i.e., formed due to activation by
the bait and not due to interaction of the bait with cDNA-encoded
proteins). As discussed below, knowledge of the frequency of Leu+
colonies that arise from activation by the bait itself will also be
important in determining the number of Leu+ colonies to pick for
further analysis during an interactor hunt.
A second important test of the activating potential of a bait is
its ability to activate the lacZ reporter. Generally, the most
sensitive lacZ reporters (e.g., plasmid pSH18-34) are not as
sensitive as the LEU2 reporters. In most cases a bait that produces
Leu+ colonies at a frequency less than
10-4 will not activate the lacZ
gene, as measured by the ability of a colony to turn blue on an X-Gal
plate. However, in rare instances and for unknown reasons, a bait
that activates a very low level of the LEU2 reporter will activate
the lacZ reporter to a significant level. Thus, it is essential to
test for activation of the lacZ reporter when characterizing the
bait.
Protocol 1 Testing whether a bait activates
transcription
____________________________________________________________________________
Reagents
- Media recipes can be found at the web site
(Finley et al., 1997) and elsewhere (Ausubel et al., 1987-1996;
Finley and Brent, 1995; Guthrie and Fink, 1991).
- Liquid YPD media
- Liquid dropout media (Glu ura-, Glu
ura-his-)
- Dropout plates (Glu ura-, Glu ura-his-, Glu
ura-his- trp-, Gal/Raf ura-his- trp-, Gal/Raf ura-his-
trp-leu-)
- X-Gal plates (Gal/Raf ura-his- trp-
X-Gal)
- Yeast strain EGY48
(MATµura3
his3 trp1 3LexAop-LEU2::leu2) or one of the less
sensitive LEU2 reporter strains EGY191 or EGY189
(MATµura3
his3 trp1 1LexAop-LEU2::leu2) (Estojak et al.,
1995)
- The URA3 2 µ lacZ reporter plasmid
pSH18-34, or a less sensitive lacZ reporter (Finley and Brent,
1995)
- HIS3 2 µ bait plasmid (e.g., a derivative
of pEG202) expressing your bait protein fused to LexA
- Two control bait plasmids: one that encodes
LexA fused to an activator like Gal4 as in the plasmid pSH17-4,
and one that encodes a transcriptionally inert bait like LexA-Max
(Zervos et al., 1993)
- The TRP1 2 µ library plasmid, pJG4-5,
lacking cDNA
- See attached transformation protocol for
additional reagents
Method
1. Construct the selection strain
either by serial transformation
of EGY48 with pSH18-34 followed by your bait plasmid, or by
co-transformation of EGY48 with your bait plasmid and pSH18-34. The
selection strain (EGY48/pSH18-34/bait plasmid) should be grown on
ura-his- medium in all subsequent steps to maintain selection for the
bait and lacZ reporter plasmids. Pick three individual transformant
colonies and streak to another Glu ura-his- plate for storage and
later recovery. All three should behave identically in the tests
below, in which case any one will serve as the selection strain into
which the library will be introduced.
2. Transform the selection strain with pJG4-5 and
select transformants on Glu
ura-his-trp- plates. Take this
opportunity to practice transforming
the selection strain at high efficiency;
this will be necessary for transformation with the library DNA
(Protocol 3).
3. Pick two or three transformant colonies and
inoculate 10 ml liquid Glu ura-his-trp- medium (again, all colonies
should behave the same, but performing the test on more than one can
help ensure that the results are not due to some rogue mutant yeast
or contaminant). Grow the liquid cultures at 30oC with shaking to
OD600=1.0 (corresponding to about 107 cells/ml). This is mid-log
phase, provided the culture started at OD600<0.2. If overnight
cultures grow to a density greater than OD600=1.0, dilute to less
than OD600=0.2 and then grow to OD600=1.0 so that the cells are in
mid-log phase when harvested.
4. Make serial dilutions from 10-1 to 10-6 of each
culture in sterile water.
5. Plate 100 ml
of the culture and 100 ml
each dilution onto two platesa:
- Gal/Raf ura-his-trp-
- Gal/Raf ura-his-trp-leu-
Incubate at 30oC.
6. Monitor the emergence of colonies during the
next several days. Calculate the number of CFU that were plated on
each Gal/Raf ura-his- trp-leu- plate by counting the number of
colonies that form on the Gal/Raf ura-his-trp- plates. Calculate the
number of Leu+ colonies/CFU. It is also worth taking note of the size
of colonies after 2, 3, and 4 days (see below).
7. Test for lacZ expression. One way to do this is
simply to patch individual transformants from step 2 to Gal/Raf
ura-his- trp- X-Gal plates (about 1 cm x 1 cm patches) and incubate
at 30oC. Yeast with a control LexA-activator fusion should turn blue
overnight while those lacking LexA or containing a transcriptionally
inert bait will remain white indefinitely. Alternatively, if the
frequency of Leu+/CFU is higher than 10-4, it may be
useful to replica plate from one of the Gal/Raf ura-his- trp-leu-
plates (one with 200-500 colonies) to Gal/Raf ura-his- trp- X-Gal.
This will reveal the frequency of blue colonies among the Leu+
colonies, a number that may be useful in determining hunt strategies
(see below).
a Galactose is used in the medium because the
actual selection will eventually be done on galactose plates to
induce expression of the activation-tagged cDNA protein. Raffinose is
added to aid yeast growth; it provides a better carbon source than
galactose alone but does not block the ability of galactose to induce
the GAL1 promoter.
____________________________________________________________________________
B. Testing baits Part 2: Does the bait protein enter the
nucleus and bind to LexA operators in the reporters, and is the
full-length fusion protein made?
There are rare reports of baits that are excluded from the yeast
nucleus; it usually possible to force these into the nucleus by
including a nuclear localization domain N-terminal to LexA. Any small
level of transcription activation by a bait could be taken as an
indication that the bait protein enters the yeast nucleus. However,
the ideal bait does not activate transcription, so another test is
needed to show that it can occupy operators in the yeast nucleus. One
simple test is the repression assay. This assay is based on the
ability of most transcriptionally inert LexA fusions to inhibit
transcription when bound to LexA operators situated between the TATA
box and the upstream activating sequence (UAS) of a reporter. The
reporter used for this test is the lacZ reporter in plasmid pJK101.
This URA3 2 µ plasmid differs from pSH18-34 in that the GAL1 UAS
is located upstream of the LexA operators. The GAL1 UAS activates the
lacZ reporter at a high level in the presence of galactose, and for
this particular derivative, it also activates at a low level in yeast
grown on glucose. Any amount of repression of the GAL UAS by a bait,
either in galactose or glucose, indicates that the bait enters the
nucleus and occupies LexA operators.
Protocol 2 The repression assay
____________________________________________________________________________
Reagents
- Liquid YPD media
- Liquid dropout media (Glu ura-, Glu
ura-his-)
- Dropout plates (Glu ura-, Glu
ura-his-)
- X-Gal plates (Glu ura-his- X-Gal, Gal/Raf
ura-his- X-Gal)
- Yeast strain EGY48 or a related
strain
- The URA3 2 µ lacZ repression assay
reporter plasmid pJK101
- HIS3 2 µ bait plasmid expressing your
bait protein fused to LexA
- Two HIS3 2 µ control bait plasmids: one
that encodes LexA fused to a transcriptionally inert protein, like
Bicoid in pRFHM1, or LexA-Max (Zervos et al., 1993), and one that
encodes no LexA, for example pRFHM0.
Method
1. Transform EGY48 with pJK101 and select
transformants on Glu ura- plates.
2. Combine three colonies from these plates and
transform them with the HIS3 bait plasmid (and the HIS3 control
plasmids). Select transformants on Glu ura-his- plates.
3. Pick four individual colonies from each
transformation and streak a patch of them onto Glu ura-his- and
Gal/Raf ura-his- plates containing X-Gal. Incubate at
30oC.
4. Examine the X-Gal plates after 1, 2, and 3
days. Yeast lacking LexA will begin to turn blue on the Gal/Raf
plates after one day and will appear light blue on the glucose plates
after two or more days. Yeast containing a bait that enters the
nucleus and binds operators will turn blue more slowly than the yeast
lacking LexA.
5. Baits that repress transcription of lacZ in
pJK101 by 2-fold or less may not cause a visible reduction in blue on
X-Gal plates. If no repression is observed on the X-Gal plates,
perform the more sensitive liquid ß-galactosidase assays with
transformants from step 2. Grow the transformants in 5 ml Glu
ura-his- and Gal/Raf ura-his- liquid media, or on Glu ura-his- and
Gal/Raf ura-his- plates for 2 days, before doing
ß-galactosidase assays (Miller, 1972).
____________________________________________________________________________
An ideal bait protein for an interactor hunt is one that does not
itself activate transcription but does repress in the repression
assay. It is also useful to verify that the full-length fusion
protein is made. In some instances, proteases in yeast will cleave
specific portions of a bait, leaving a truncated LexA fusion that
still binds to operators. To demonstrate that the full-length bait
protein is made one can perform a Western blot on extracts from yeast
cells that harbor the bait plasmid, immunoblotting with either an
antibody to LexA or one specific to the protein fused to LexA. The
simplest way to do this is to prepare yeast cell extracts by growing
yeast in liquid culture (lacking histidine to maintain selection for
the bait plasmid) to OD600 = 0.5, spinning 1
ml of the
culture to pellet the cells, and resuspending the cells in 50
ml of 2X Laemmli sample buffer (Laemmli,
1970). The cells can then be broken by freezing on dry ice followed
by boiling for 5 min prior to loading on an SDS polyacrylamide gel
(about 15 ml/lane). The proteins can then
be transferred to a filter and blotted with standard immunoblotting
(Western) methods (Ausubel et al., 1987-1996; Harlow and Lane,
1988).
C. Screening a library for interactors
Most cDNA libraries available for the Brent lab version of the
yeast two-hybrid system contain over
106 individual cDNAs (in plasmid
pJG4-5). In theory, a library with
106 individual cDNAs includes cDNAs
for messages that were more frequent than 1 in
106 mRNA molecules in the mRNA
population used to make the library. To have a chance at isolating
the rarest cDNAs in a library, it is important to collect more yeast
transformants than there are individual cDNAs in the library. Thus,
for a library with 106 individual
cDNAs, one might try to obtain 2-3 x
106 yeast transformants. With the
most common yeast two-hybrid strains one can obtain up to
105 transformants per µg of
library plasmid DNA using the attached transformation protocol.
A pilot transformation should be performed with the selection
strain to determine the transformation efficiency that can be
obtained. This allows one to calculate how many individual
transformations to set up to obtain the desired number of total
transformants. The transformation mixes are plated onto 22cm x 22cm
Glu ura-his-trp- plates, attempting to get 1-2 x
105 transformants/plate. Again, the
number of individual transformation mixes to put on each plate is
calculated from the expected transformation efficiency derived from
pilot experiments. The transformants are collected and stored frozen.
Aliquots are then plated to ura-his-trp-leu- Gal/Raf plates to select
interactors.
Protocol 3 Transforming the selection strain and selecting
potential interactors
____________________________________________________________________________
Reagents
- Liquid dropout media (Glu ura-his-, Gal/Raf
ura-his-trp-)
- Dropout plates (Glu ura-his-trp-, Gal/Raf
ura-his-trp-leu-, Glu ura-his-trp-leu-)
- X-Gal plates (Glu ura-his-trp- X-Gal, Gal/Raf
ura-his-trp- X-Gal)
- Sterile water
- Sterile glycerol solution (65% (v/v) glycerol,
0.1 M MgSO4, 25 mM Tris-HCl 7.4).
- Glass beads (4 mm diameter; Fisher
Scientific), sterilized by autoclaving.
- Sterile 50 ml Falcon tubes
- Sterile 50 ml round-bottom polypropylene
centrifuge tubes
Method
1. Using the selection strain prepared in Protocol
1, perform pilot transformations (as suggested in Protocol 1 step 2)
to determine transformation efficiency.
2. Based on your transformation efficiency,
calculate the number of transformations to obtain the desired number
of total transformants (i.e., each transformation = 1 µg library
DNA/50 µl of cells as described in the transformation protocol).
Also, calculate the number of transformations to be plated on each
22cm x 22cm Glu ura-his-trp- plate to get 1-2 x 105
transformants/plate (e.g., if your efficiency in pilot experiments is
5 x 104 transformants/µg you should set up 2
transformations for each 22cm x 22cm plate).
3. Based on the above calculations, grow the
appropriate amount of the selection strain in liquid Glu ura-his-
medium and set up the necessary number of transformations (see
attached transformation protocol).
4. After the heat shock, invert the tubes several
times to mix - gently. Remove 10 µl from several of the
transformation mixes and make three dilutions (10-1,
10-2 and 10-3) each in sterile water. Plate 100
ml of
each dilution onto 100 mm diameter Glu ura-his-trp- plates and
incubate at 30oC. This will allow the total number of transformants
to be calculated.
5. Plate the remainder of the transformation mixes
(less then 2 ml total/plate) onto 24cm X 24cm Glu ura-his-trp- plate.
There is no need to spin the cells or remove the PEG. The medium in
these plates should be at least 0.6 cm thick, level, and free of
bubbles. To achieve an even distribution of cells, pour about 100
sterile glass beads (4 mm diameter) onto the plate with the cells.
Gently roll the beads around the plate to distribute the
transformation mix, then pour the beads off, or onto the next plate.
This technique works best when the surface of the plates is not too
wet so that the medium absorbs the transformation mix. To achieve
this moisture content, put newly solidified plates into a laminar
flow hood with the lids ajar for about 1 h before plating.
6. Incubate the plates at 30oC. Colonies should
appear after about 24 h. Continue incubation until colonies are 1 - 2
mm in diameter, which should take a total of approximately 2
days.
7. Place the plates at 4oC for 2 - 4 hours to
harden the agar. Using the long edge of a sterile 75mm x 50mm glass
microscope slide (and sterile technique!), scrape the yeast from the
plate. Try not to scrape any agar as this will interfere with
pipetting. Collect the yeast from the glass slide by wiping it on the
lip of a sterile 50 ml Falcon tube.
8. Wash the cells twice with 2 or 3 volumes of
sterile TE. It may be necessary to split into two or more tubes to
effectively pellet. It is best to pellet the cells each time in a
sterile round bottom polypropylene tube at 2000 g for 4 min so they
may be easily resuspended. The pellet volume for 500,000
transformants will be about 8 ml.
9. Resuspend the cells thoroughly by swirling in 1
pellet volume of sterile glycerol solution. Mix well by
vortexing on low speed. Freeze 1 ml aliquots at -70oC.
10. Determine the plating efficiency by thawing an
aliquot of library transformants and making serial dilutions in
sterile water. Plate 100 ml
of each dilution onto 100 mm diameter Gal/Raf ura-his-trp- plates.
Count the colonies that grow after 2 - 3 days at 30oC. Represent the
plating efficiency in colony forming units (CFU)
per unit volume of frozen cells. Note: to save time one can estimate
the plating efficiency as ~108 CFU/100 ml,
and immediately proceed to steps 11 and 12. Once the actual plating
efficiency is known, calculate the number of CFU that were actually
plated in steps 11 and 12.
11. Thaw a 1 ml aliquot of transformed yeast and
dilute 10-fold into 9 ml Gal/Raf ura-his-trp- liquid medium. Incubate
at 30oC with shaking for 6 to 8 h to induce the GAL1 promoter and
expression of the library encoded proteins. Pellet the cells by
centrifugation at 2000 g for 4 min at 20 - 25oC and resuspend in 10
ml sterile water.
13. Plate less than 106 CFU (determined from the
plating efficiency test in step 10) onto each 100 mm diameter Gal/Raf
ura-his-trp-leu- plates. To avoid overcrowding of Leu+ colonies, do
not plate more CFU than are expected to produce ~20 background
Leu+/plate (as determined in Protocol 1). Incubate the selection
plates at 30oC. Colonies should appear in 2 - 5 days. To keep the
plates from drying out after two days, it may be helpful to put them
in plastic bags or containers, or put parafilm around each
plate.
14. Pick colonies (see discussion below for number
to pick) with sterile toothpicks or applicator sticks and patch, or
streak for single colonies, onto another Gal/Raf ura-his-trp-leu-
plate. If the Leu+ colonies are closely spaced it will be necessary
to streak purify to single colonies to separate the different Leu+
clones. Ideally the Leu+ yeast should be streaked for single colonies
to isolate them from contaminating Leu- yeast. However, when there
are large numbers of Leu+ colonies to pick, it may be inconvenient to
streak purify every one; in this case, growing patches on a second
selection plate will at least enrich for the Leu+ cells.
15. To show that the Leu+ phenotype is
galactose-dependent, patch (or replica plate) the Leu+ yeast onto Glu
ura-his-trp- master plates to turn off the GAL1
promoter and stop expression of the activation-tagged cDNA protein.
Grow at 30oC for about 24 h.
16. Replica the master plates to the following
five plates, in order: 1. Gal/Raf ura-his-trp- X-Gal; 2. Glu
ura-his-trp- X-Gal; 3. Glu ura-his-trp-leu-; 4. Gal/Raf
ura-his-trp-leu-; 5. Glu ura-his-trp-. Incubate at 30oC and examine
the results after 1, 2, and 3 days.
17. Pick only those yeast that are Leu+ on
galactose but not glucose. Keep in mind that if Leu+ clones were not
purified in step 14, some patches may be contaminated with background
Leu+ yeast, which will not be galactose-dependent. The
galactose-dependent Leu+ phenotype indicates that reporter activation
depends on expression of the library protein. Further characterize
these by isolating the library plasmid and determining the
interaction specificity.
____________________________________________________________________________
Alternate protocol - liquid selection and amplification of
Trp+ library transformants. We have had some success at
selecting and amplifying library transformants in liquid culture (M.
Kolonin and R. Finley, unpublished). To do this, we dilute individual
transformation mixes after heat shock (from Protocol 3 step 4)
50-fold into liquid Glu ura-his-trp- medium and grow shaking at
30oC until the OD600 is
~2.0. The OD600 of this culture
begins at less than 0.2 and usually takes 30-48 hours to reach 2.0.
We then harvest the cells and proceed as in Protocol 3 step 8. By
removing aliquots immediately after dilution and before harvesting
and plating on Gal/Raf-ura-his-trp- we have estimated that
transformants are amplified approximately 100-fold in this procedure.
This approach eliminates the cost and inconvenience of selecting
transformants on plates. The disadvantage is that there is no
reliable way to verify that library transformants are evenly
amplified.
How many Leu+ colonies should be picked? When
considering how many Leu+ colonies to pick at step 14 of Protocol 3,
it is important to take into account the background frequency of Leu+
colonies that the bait itself produces (represented as Leu+
colonies/CFU), as determined in Protocol 1, and the total number of
library transformants obtained. To completely screen all of the
library transformants, the minimum number of Leu+ colonies one would
need to pick and characterize can be estimated by:
# to pick > (# Leu+ colonies/CFU) X (total # of library
transformants)
If, for example, the background for a given bait were
10-5 Leu+ colonies/CFU, one would
need to pick and characterize at least 10 colonies to screen through
106 library transformants. More to
the point, the first 10 colonies picked would be expected to be
background, so to get an interactor that is rare in the library one
might need to pick and characterize 20 or 30 Leu+ colonies.
Should galactose-dependent Leu+ colonies that do not turn
blue on the X-Gal plates be further characterized? Yes. Of
the galactose-dependent positives, several different classes of Leu
and lacZ phenotype are possible. For example:
Class I. galactose-dependent Leu+ galactose-dependent dark blue on
X-Gal
Class II. galactose-dependent Leu+ galactose-dependent light blue
on X-Gal
Class III. galactose-dependent Leu+ white on X-Gal
Many hunts will yield Leu+ colonies from each class. Often this
indicates that at least three different interactors are represented
among the positives. A common mistake is to concentrate on only the
"strongest" class (Class I above) and ignore the "weaker" class
(Class III) which can include biologically significant interactors
(Finley et al., 1996).
The next step for the galactose-dependent positives is to isolate
the library plasmid from each and re-introduce it into the selection
strain to show that the putative interaction phenotype depends on the
library plasmid and not on mutations in the yeast or reporter genes.
This test can often be performed at the same time as the specificity
test described below. If the library has been properly screened to
exhaustion, each interactor cDNA should be represented more than once
in the putative positives. cDNAs corresponding to abundant messages
may have been isolated many times. To reduce the amount of work in
subsequent steps it is useful to determine which yeast contain
identical cDNAs. This can be easily done by performing PCR with
primers flanking the cDNA insertion site using DNA template from a
quick yeast miniprep (Finley and Brent, 1995). PCR products can be
digested with HaeIII and AluI and run on an agarose gel to reveal
unique restriction fragment patterns for each cDNA (Finley and Brent,
1995). One or two of each unique library plasmid can then be rescued
in E.coli and used in the specificity test.
D. Determining the specificity of interactors
Many of the proteins identified in interactor hunts are
non-specific interactors: they appear to interact with a number of
different unrelated LexA fusions. Non-specific interactors are
frequently isolated in hunts using unrelated baits. They can be
identified and discarded by testing the ability of the cDNA-encoded
proteins to interact with a handful of bait proteins unrelated to the
original bait. cDNA-encoded proteins that interact only with the
original bait and not with unrelated baits are considered true
specific interactors. The specificity test can be performed by
introducing rescued library plasmids into different selection strains
that each harbor a different bait plasmid. Transformants are picked
and patched onto a Glu ura-his-trp- plate and then replica plated to
indicator plates as in Protocol 2 steps 15 and 16. This method of
testing specificity can be somewhat cumbersome if a large number of
different library plasmids were isolated, and if these are to be
tested for interaction with several different baits. For this reason
we use the interaction mating assay (Finley and Brent, 1994) to
perform the specificity test, as described in Protocol 3.
Interestingly, the commonly isolated non-specific interactors,
which include heat shock proteins, ribosomal proteins, proteasome
subunits, and other proteins, are not isolated in every interactor
hunt, and in fact do not appear to interact with every bait. This
highlights the importance of using several different bait proteins to
test the specificity of an interactor. For example, frequently a
non-specific interactor will interact with just 30% of the bait
proteins tested. If only one or a few bait proteins are tested, a
non-specific interactor could appear to be specific.
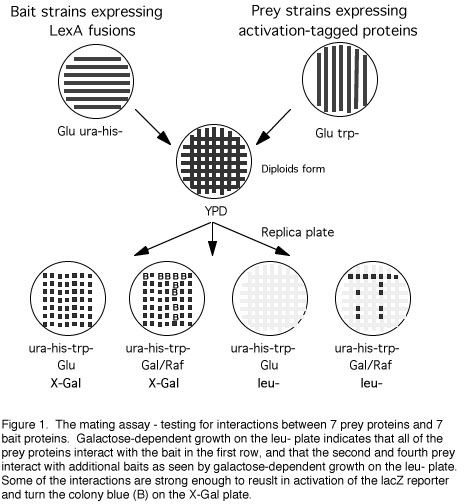
Protocol 4 The interaction mating assay
____________________________________________________________________________
Equipment and Reagents
- Rescued library plasmid DNA
- Liquid YPD medium
- Liquid dropout media (Glu ura-)
- YPD plates
- Dropout plates (Glu trp-, Glu ura-his-, Glu
ura-his-trp-, Gal/Raf ura-his-trp-leu-, Glu
ura-his-trp-leu-)
- X-Gal plates (Glu ura-his-trp- X-Gal, Gal/Raf
ura-his-trp- X-Gal)
- Applicator sticks (e.g. FisherBrand 01-340),
or toothpicks, sterilized by autoclaving.
- Replica plating apparatus and sterile velvets
or filters.
- Yeast strain RFY231
(MATa
ura3his3 leu2::3LexAop-LEU2 trp1::hisG
LYS2) or EGY48. Note: RFY231 is EGY48 with the trp1-1
allele deleted (R. Finley, unpublished).
- Bait strains: S. cerevisiae strain RFY206
(MATa ura3-52 his3Æ200 leu2-3 lys2Æ201
trp1::hisG) transformed with a URA3 plasmid containing a lacZ
reporter, such as pSH18-34, and various HIS3 bait plasmids, such
as derivatives of pEG202 that produce different LexA fusions. Each
bait strain will contain a different bait plasmid. One strain
should contain the original bait used in the interactor
hunt.
Method
1. Transform yeast strain RFY231 with the rescued
TRP1 library plasmids and select transformants on Glu trp- plates (if
EGY48 is substituted for RFY231, more than one Trp+ transformant
should be analyzed to ensure than a trp1-1 revertant has not
been selected). As a control, transform RFY231 with a library plasmid
pJG4-5 that has no cDNA insert.
2. Use sterile applicator sticks or toothpicks to
streak individual RFY231 transformants onto standard 100 mm Glu trp-
plates in parallel lines (see Figure 1). Streaks should be at least 3
mm wide and at least 5 mm apart, with the first streak starting about
15 mm from the edge of the plate. A 100 mm plate will hold up to 8
different bait strains. Include at least one streak of the
transformants with the control plasmid (no cDNA). Create a duplicate
plate of streaked RFY231 transformants for each plate of bait strains
to be used.
3. Likewise, streak different bait strains in
vertical parallel stripes on a Glu ura-his- plate. Create a duplicate
plate of bait strains for each different plate of prey strains to be
used. Incubate both sets of plates at 30oC until growth is heavy.
When taken from reasonably fresh cultures (for example, plates that
have been stored at 4oC for less than a month) streaked
RFY206-derived bait strains take about 48 hours to grow and
RFY231-derived strains take about 24 hours.
4. Print the RFY231 derivatives and the RFY206
derivatives onto the same replica filter or velvet so that the
streaks from the two plates are perpendicular to each other (see
Figure 1).
5. Lift the print of the two strains from the
filter or velvet with a YPD plate. Incubate the YPD plate at 30oC
overnight. Diploids will form where the two strains intersect. One
strain may grow more rapidly than the other during this time but this
does not hinder formation of diploids in the
intersections.
6. Replica from the YPD plate to the following
indicator plates, in order: 1. Gal/Raf ura-his-trp- X-Gal; 2. Glu
ura-his-trp- X-Gal; 3. Glu ura-his-trp-leu-; 4. Gal/Raf
ura-his-trp-leu-; 5. Glu ura-his-trp-. Incubate at 30oC and examine
the results after 1, 2, and 3 days. Only diploids will grow on the
X-Gal plates and only interactors will grow on galactose plates
lacking leucine (Figure 1).
____________________________________________________________________________
What next? Although the methods described above
allow several types of false positive to be eliminated, they do not
address the biological significance of the interactions observed. In
some instances the sequence of a specific interactor will suggest
that its interaction with the bait may have a real in vivo
function. However, two-hybrid interactions can occur between proteins
that normally do not interact (for example, because they are never
expressed at the same time or in the same tissue or subcellular
compartment). A good first step to show biological significance is to
verify the interaction by a different, biochemical technique,
preferably co-precipitation from a cell in which both proteins are
expressed. Ideally, the next step would involve a functional assay
for the new protein, to show, for example, that the new protein is
involved in the same biological process as the bait protein. The
following two sections include a few additional ways to address
function.
IV.
TWO-HYBRID METHODS TO STUDY LARGE SETS OF PROTEINS AND PROTEIN
NETWORKS
Finding interacting partners can reveal much about the function of
a protein. Most regulatory proteins, for example, appear to function
by contacting other proteins. This is true for proteins that regulate
many different cellular processes, including transcription,
translation, DNA replication, signal transduction, cell cycling,
differentiation, and programmed cell death. All of the proteins
involved in a given process together can be thought of as a network
of interacting proteins. The members of each interacting network are
linked through protein-protein contacts. A complete understanding of
any given process can only be achieved when all of the components of
the protein network regulating it are identified. Yeast two-hybrid
systems offer approaches to characterizing individual interactions
and whole networks of proteins.
Isolating a new interacting protein can reveal information about
function if the sequence of the new interactor indicates similarity
or identity with a protein whose function has been at least partially
characterized. However, it is still often the case that the sequence
of a interacting protein reveals little about its function. Another
approach is to assume that the new protein functions in the same
network as the original bait protein and to use the new protein as a
bait to identify other members of the network. Repeating this process
increases the chances of isolating a previously characterized
protein, or one whose sequence provides clues to function. In
principle, this approach could be used repeatedly to isolate all of
the components of a regulatory network. Because some regulatory
proteins may be shared by different cellular processes (e.g.
regulation of cell cycle and DNA replication by p21CIP1 (Li et
al., 1994)), and networks for many different processes may be
connected (e.g. a signal transduction pathway and the activation of
gene transcription), this approach could identify many expressed
genes from a small number of starting points.
An approach complementary to performing sequential hunts is to use
the interaction mating assay to look for interactions between
increasingly large sets of proteins (Bartel et al., 1996; Finley and
Brent, 1996). In one variation of this approach, large panels of
baits are collected in baits strains placed on plates in grids (e.g.,
in the standard 96-well format). The grids can then be screened
simultaneously for interactions with individual prey proteins. Bait
strains can be created as described in Protocol 4 using bait plasmids
that express various proteins of known and unknown function. Large
panels of bait strains can be collected and stored frozen
indefinitely and then screened against any number of prey
strains.
One such collection contains over 700 different bait proteins from
our own work and from numerous other labs that use the interaction
trap. Screening a protein against such a panel enables one to quickly
test its ability to interact with a large number of known proteins,
most of which have been characterized to some extent, and have been
chosen for study because of their known or suspected involvement in
some biological process. Thus, finding an interaction between a
tested protein and a member of the panel often gives an immediate
clue about the biological function of both proteins. While the number
of proteins in any such panel is far less than the number of proteins
in a good library, this approach does offer the advantage of
screening the test protein against a set of proteins enriched for
those of current interest to the biological community. More
restricted panels of bait proteins, for example those known or
suspected to function in a particular pathway, or those isolated in
sequential interactor hunts, can provide a useful resource for
characterizing new proteins. Such a panel may also be useful to
characterize differences in the patterns of interactions made by
wild-type and mutant variants of proteins such as those created in
vitro or associated with particular diseases or other phenotypes.
For some proteins, this approach offers additional advantages over
screening a library using a traditional two-hybrid scheme. Proteins
that activate transcription when fused to LexA or another DNA-binding
domain can be difficult to use in conventional interactor hunts.
Though methods are available to reduce the sensitivity of the
reporter genes (Durfee et al., 1993; Estojak et al., 1995) it is not
always possible to reduce the reporter sensitivity below the
threshold of activation for some baits. Moreover, reduction in
reporter sensitivity carries with it the risk that the reporters will
not detect weakly interacting proteins. Thus, an alternative for
proteins that activate transcription as baits, is to use them as
preys to screen existing panels of baits, or even libraries of baits.
Interaction mating approaches also have clear advantages for proteins
that are somewhat toxic to yeast; the prey vector allows conditional
expression of toxic proteins in the presence of a bait, and often the
interaction can be observed because the reporters are activated even
if the cells subsequently become inviable.
V.
TESTING THE FUNCTION OF INDIVIDUAL INTERACTIONS
Finding the position of a protein within a network of interacting
proteins can provide information about the function of the protein
and the network. However, ultimately, the nature of each individual
protein-protein contact must be understood. Several two-hybrid
methods allow the significance of individual protein interactions to
be analyzed.
A. Mapping interaction domains
Determining the domains within a protein that are responsible for
its interaction with other proteins can provide a valuable insight
into the way a protein functions. Several approaches are available to
map interaction domains with yeast two-hybrid methods. All start with
a bait protein and prey protein that interact and activate the
reporter genes. Derivatives of one of these proteins are constructed
and tested for interaction with the other. We usually make
derivatives of the prey protein because derivatives of the bait
protein may differ in their ability to activate the reporters by
themselves, which complicates interpretation of the results.
Derivatives of the prey protein can be made and tested for
interaction with the bait in several ways. In any approach it is
important to keep in mind that the prey is a fusion to an N-terminal
activation domain and must be maintained in the correct reading
frame. One approach is to subclone restriction fragments encoding
parts of the prey fusion protein into the prey vector (i.e., pJG4-5
or derivative) and introduce the resulting vectors individually into
selection strains harboring the bait vector or control vectors.
Alternatively, derivatives can be tested for interaction using the
mating assay as described in Protocol 4. A second approach is to make
N-terminal or C-terminal deletion derivatives of the prey fusion
protein and test them for interaction with the bait, again by
individual transformation into selection strains or by the mating
assay. Deletion derivatives can be constructed in a cloning vector
(Ausubel et al., 1987-1996), and then subcloned into the prey vector,
pJG4-5. Alternatively, the deletion derivatives can be constructed
directly in a derivative of the prey vector. For example, pZP4-5o and
pJF3 are derivatives of pJG4-5 that have unique, rare restriction
sites downstream of the cDNA cloning sites which allow C-terminal
deletions to be constructed by unidirectional exonuclease III
digestion from the 3' end of the insert (R. Finley, Z. Paroush, and
J. Fonfara, unpublished). Similarly, pJF2 contains unique 5'
restriction sites that allow N-terminal deletions to be constructed.
A third approach is to make random DNA fragments encoding parts of
the prey protein, for example by sonication (e.g., ref. (Stagljar et
al., 1996), and insert these into the prey vector. Finally, a variety
of techniques are available to make single and multiple point
mutations of one interactor, which can then be inserted into the prey
vector to test for interaction with a bait.
B. Construction of dominant negative mutants
A powerful approach to understanding protein function is to create
and express dominant negative forms of the protein that inactivate
the function of the wild-type version (Herskowitz, 1987). The yeast
two-hybrid system provides a method to design and assay potential
dominant negatives. One type of dominant negative is a protein
mutated so that it still interacts with one of its protein partners
but lacks other functional domains. In this case the "partner" could
be another protein or the same protein if it forms homodimers.
Expression of the mutant form of the protein might be expected to
bind to the partner protein and make it inaccessible to the wild-type
version. One way to create such a mutant is to isolate the minimal
domain of a protein that will interact with another protein partner
as described in the previous section. If the interacting domain is
just a fraction of the protein it would be expected to lack other
functional domains, and would therefore be a candidate dominant
negative. A related but more precise approach could be used for
proteins that have at least two different known partners. For
example, if protein A interacts with both proteins B and C, mutant
varieties of protein A could be constructed and tested in the
two-hybrid assay for their ability to interact with just protein B
but not protein C. In this case, we would have precise knowledge of
the function missing in the dominant negative (interaction with
protein C).
It is worth noting that, while the dominant negative effect is
frequently open to multiple interpretations (Herskowitz, 1987),
functional inferences from the type of dominant negatives referred to
here may be less uncertain. This is because we know that the dominant
negative interferes with a specific protein interaction; we have
designed it that way and tested it in the two-hybrid system.
C. Disrupting protein interactions
The yeast two-hybrid system provides an assay to develop reagents
that disrupt protein interactions. Such reagents can be used in
vivo to probe the function of individual protein interactions.
Frequently a protein makes functional contacts with several other
proteins. For example, the catalytic subunit of a protein kinase may
interact with one or more regulatory subunits and with substrates.
Deletion of the gene encoding the kinase could provide information
about the function of the protein as a whole, but would not provide
information about the individual interactions that it makes with
other proteins. As mentioned in the previous section, certain types
of dominant negative mutants may be created that interfere with
specific interactions made by a wild-type protein. In the kinase
example, a dominant negative kinase might be created that interacts
with its regulatory subunit but not its substrate; such a mutant
would be expected to compete with the wild-type kinase for regulatory
subunits.
Another type of potential disrupter of protein interactions that
can be identified with the two-hybrid system is a peptide that
interacts tightly and specifically with one of a pair of interacting
proteins. Such peptides have been isolated from a random peptide
library using the interaction trap yeast two-hybrid system as
described by Colas et al. (Colas et al., 1996). These authors created
a peptide library using a plasmid related to pJG4-5 that expressed
random peptides fused to an activation domain and an inert platform
molecule, E.coli thioredoxin. To find peptides that interact
specifically with a bait protein an interactor hunt is performed as
described in Protocol 2. Some of the specific peptides, called
aptamers, would be expected to interact with surfaces
of the bait that are required for interactions with other proteins.
These are potential disrupters of specific protein interactions.
A two-hybrid assay can also be used to show that a potential disrupter can interfere with a protein-protein interaction. The two proteins can be expressed, one as a bait and one as a prey, and then the potential disrupter can be expressed to see if it reduces the ability of the bait and prey to interact and activate a reporter. We developed a method to test whether an interacting domain or a peptide aptamer can disrupt specific interactions (M. Kolonin and R. Finley, unpublished). A potential disrupter is first isolated as an interactor. The library plasmid expressing the potential disrupter is isolated and used to transform RFY231, and these transformants are mated with a special bait-prey interaction strain as described in Protocol 4. In this case, however, the bait strain expresses the original bait as a prey (activation domain fusion) from plasmid pMK1, and a protein that interacts with it as a bait. Disruption of the interaction results in loss of LEU2 transcription and inability to grow on leu- plates.
The methods outlined here present an integrated approach to
understanding the function of proteins, protein interactions, and
networks of proteins. First, all of the potential partners of a
protein thought to be involved in a particular biological process can
be identified. Second, many additional members of the same regulatory
network can be identified in successive interactor hunts. Third,
interaction domains can be mapped. Fourth, mutants incapable of
specific interactions can be identified, and in many cases these
mutants can be expressed in vivo to provide functional
information. Finally, reagents can readily be developed that disrupt
specific protein interactions, and then can be used to probe the
function of these interactions in vivo.
Acknowledgments
I thank Mikhail Kolonin, Jennifer Fonfara, and Catherine Nelson,
for providing comments, and Mikhail Kolonin and members of the Finley
lab for contributions to the protocols. I also thank the members of
the Brent lab for their many contributions to the protocols. I
especially thank Roger Brent who co-wrote previous versions of the
interactor hunt protocols.
References
Ausubel, F. M., Brent, R., Kingston, R. E., Morre,
D., Seidman, J. G., and Struhl, K. (1987-1996). Current protocols in
molecular biology (New York: Greene and
Wiley-interscience).
Bartel, P. L., Roecklein, J. A., SenGupta, D., and
Fields, S. (1996). A protein linkage map of Escherichia coli
bacteriophage T7. Nature Genetics 12, 72-77.
Brent, R., and al., e. (1997).
http://xanadu.mgh.harvard.edu/brentlabhomepage4.html. web
site.
Colas, P., Cohen, B., Jessen, T., Grishina, I.,
McCoy, J., and Brent, R. (1996). Genetic selection of peptide
aptamers that recognize and inhibit cyclin- dependent kinase 2.
Nature 380, 548-550.
Durfee, T., Becherer, K., Chen, P.-L., Yeh, S.-H.,
Yang, Y., Kilburn, A. E., Lee, W.-H., and Elledge, S. J. (1993). The
retinoblastoma protein associates with the protein phophatase type 1
catalytic subunit. Genes and Dev. 7, 555-569.
Estojak, J., Brent, R., and Golemis, E. A. (1995).
Correlation of two-hybrid affinity data with in vitro measurements.
Mol Cell Biol 15, 5820-5829.
Finley, R. L., Jr., and Brent, R. (1994).
Interaction mating reveals binary and ternary connections between
Drosophila cell cycle regulators. Proc Natl Acad Sci U S A 91,
12980-12984.
Finley, R. L., Jr., and Brent, R. (1995).
Interaction trap cloning with yeast. In DNA Cloning, Expression
Systems: A Practical Approach, B. D. Hames and D. M. Glover, eds.
(Oxford: Oxford University Press), pp. 169-203.
Finley, R. L., Jr., and Brent, R. (1996).
Two-hybrid analysis of genetic regulatory networks. In The yeast
two-hybrid system, P. L. Bartel and S. Fields, eds. (Oxford: Oxford
University Press).
Finley, R. L., Jr., Thomas, B. J., Zipursky, S.
L., and Brent, R. (1996). Isolation of Drosophila cyclin D, a protein
expressed in the morphogenetic furrow before entry into S phase.
Proc. Natl. Acad. Sci. USA 93, 3011-3015.
Finley, R. L. J., and al., e. (1997).
http://cmmg.biosci.wayne.edu/rfinley/finlab/finlab-home.html. web
site.
Guthrie, C., and Fink, G. R. (1991). Guide to
yeast genetics and molecular biology. In Methods in enzymology
(Boston: Academic Press, Inc.).
Gyuris, J., Golemis, E., Chertkov, H., and Brent,
R. (1993). Cdi1, a human G1 and S phase protein phosphatase that
associates with Cdk2. Cell 75, 791-803.
Harlow, E., and Lane, D. (1988). Immunoblotting.
In Antibodies: A laboratory manual (Cold Spring Harbor, N.Y.: Cold
Spring Harbor Laboratory), pp. 471-510.
Herskowitz, I. (1987). Functional inactivation of
genes by dominant negative mutations. Nature 329,
219-22.
Laemmli, U. K. (1970). Cleavage of structural
proteins during the assembly of the head of bacteriophage T4. Nature
227, 680-685.
Li, R., Waga, S., Hannon, G. J., Beach, D., and
Stillman, B. (1994). Differential effects by the p21 CDK inhibitor on
PCNA-dependent DNA replication. Nature 371,
534-537.
Mendelsohn, A. R., and Brent, R. (1994).
Applications of interaction traps/two-hybrid systems to biotechnology
research. Curr. Op. Biotechn. 5, 482-486.
Miller, J. (1972). Experiments in molecular
genetics (Cold Spring Harbor, N.Y.: Cold Spring Harbor
Laboratory).
Stagljar, I., Bourquin, J. P., and Schaffner, W.
(1996). Use of the two-hybrid system and random sonicated DNA to
identify the interaction domain of a protein. Biotechniques
21, 430-432.
Zervos, A. S., Gyuris, J., and Brent, R. (1993).
Mxi1, a protein that specifically interacts with Max to bind Myc-Max
recognition sites. Cell 72, 223-32.
|
